I-клеточная болезнь
| I-клеточная болезнь | |
|---|---|
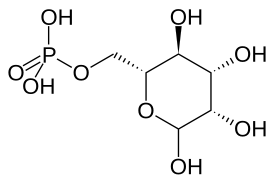 Маннозо-6-фосфат (M6P): проявления I-клеточной болезни связаны с невозможностью фосфорилирования M6P в составе гликопротеинов. | |
| МКБ-10 | E77.0 |
| МКБ-10-КМ | E77.0 |
| МКБ-9 | 272.7 |
| OMIM | 252500 |
| DiseasesDB | 29175 |
| eMedicine | ped/1150 |
| MeSH | D009081 |
I-кле́точная боле́знь (муколипидо́з II) — наследственное заболевание из группы муколипидозов, относящееся к лизосомным болезням накопления. Клиническая картина развивается в результате дефекта фосфотрансферазы (фермента аппарата Гольджи). Метаболическая роль этого фермента, принимающего участие в посттрансляционном синтезе олигосахаридной части лизосомных ферментов, заключается в синтезе специфичной метки катаболических ферментов лизосом, расщепляющих олигосахариды, липиды и гликозаминогликаны внутри клетки.
Содержание
Наследование

I-клеточная болезнь наследуется, как и подавляющее большинство лизосомных болезней накопления, по аутосомно-рецессивному типу. С одинаковой частотой встречается как у мужчин, так и у женщин. Развитие болезни связано с дефектом посттрансляционного процессинга ферментов лизосом.
Патогенез
Клиническая картина заболевания, как и в случае муколипидоза III (псевдополидистрофии Гурлер), обусловлена генетическим дефектом гена GNPTAB, расположенного на длинном плече 12-й хромосомы (12q23.3), кодирующего гликопротеин-GlcNaCI-1-фосфотрансферазу (GlcNAc-1-фосфотрансеразу). Данный фермент аппарата Гольджи фосфорилирует N-концевой остаток маннозы в маннозо-6-фосфат (М6Ф) гликопротеинов. В физиологических условиях безошибочная работа системы дифференцировки лизосомных гидролаз и отправки их в эндолизосомы возможна благодаря тому, что маннозофосфатные группы добавляются в аппарате Гольджи только к определённым гликопротеинам. Таким образом, необходимо специфическое распознавание гидролаз ферментами аппарата Гольджи, ответственными за присоединение М6Ф. В связи с тем, что все гликопротеины, которые попадают в компартмент Гольджи, обладают одинаковыми N-связанными олигосахаридными цепями, сигнал для добавления к олигосахариду М6Ф должен находиться в само́й полипептидной цепи каждой гидролазы. Для присоединения маннозофосфатных групп к лизосомным гидролазам необходима последовательная работа двух ферментов. Сначала N-ацетилглюкозамин-фосфотрансфераза (GlcNAc-фосфотрансфераза) присоединяет P-GlcNAc к 6-му углеродному атому остатка маннозы N-связанного с лизосомным гликопротеином-предшественником олигосахарида. Затем второй фермент (фосфогликозидаза) удаляет концевой фрагмент GlcNAc, оставляя фосфат — в результате образуется маннозо-6-фосфатный маркер. При этом GlcNAc-фосфотрансфераза специфически активируется сигнальным участком на лизосомных гидролазах, а фосфогликозидаза является неспецифическим ферментом. Подобная модификация некоторых остатков маннозы в г/г/с-компартменте Гольджи защищает их от возможного последующего действия маннозидаз, активных в промежуточном компартменте аппарата Гольджи. Без переноса остатка фосфорной кислоты на маннозу олигосахарида гликопротеинов катаболические ферменты транспортируются из аппарата Гольджи в межклеточное пространство. Лизосомы не могут функционировать без катаболических ферментов, которые необходимы для полноценного расщепления макромолекул (олигосахаридов, липидов, гликозаминогликанов), которые накапливаются внутри клеток организма. В результате накопления этих веществ в лизосомах формируются характерные «I-клетки» (отсюда и название заболевания) или «внутриклеточные включения». Наличие этих клеток может быть выявлено и идентифицировано в процессе микроскопии. Кроме того, неполноценные катаболические ферменты лизосом (обычно присутствующие только внутри лизосом) обнаруживаются в периферической крови.
Клиническая картина
Муколипидоз II начинается в раннем возрасте и проявляется отставанием психического развития на фоне типичного для мукополисахаридозов фенотипа. Отличительными особенностями являются отчётливые включения в культивируемых фибробластах кожи и резко повышенный уровень катаболических ферментов лизосом в периферической крови.